


Due Diligence and Quality Systems Integration… More Important than Ever during Tumultuous Period for Pharmaceutical and Medical Device Industry

Global Trade Wars … Supply Chain Interruptions … U.S. government initiatives for reshoring of manufacturing for critical Medical Device and Pharmaceutical products … these are all significant drivers for mergers and acquisitions during this period which many would say is the most tumultuous period in history for the Pharmaceutical and Medical Device Industries.
M&A and new contract manufacturing activities are at an all-time high. Hence, due diligence and Quality Systems Integrations efforts need to be efficient, with high quality outputs more than ever for Medical Device companies. There is less room for error, and it can become a matter of survival should keen competitors move faster and make the right investments while others stay behind and thereby adversely affect their product pricing, quality, and/or market share.
It is important to know the differences between Due Diligence (DD) and Quality Systems Integration (QSI) and where they may overlap.
Due Diligence (DD)
DD is a collaboration with business development and other functions to assess a target to identify potential fit for a strategic alliance, often resulting in acquisition of the organization. They often involve a dozen or more stakeholders representing different functions of the business (ex. Marketing, R&D, Clinical, Regulatory, Commercial, Supply Chain, Facilities, Operations, Quality, HR, Finance, Legal, IP, IT, etc.) all with different interest and objectives for their aspect of the assessment.
The Quality and Regulatory functions focus during DD is the identification and estimation of vulnerability risks associated with the target acquisition’s product(s) quality & regulatory compliance. Identifying these key risks can impact price negotiation, reservation of acquisition post-merger funds required for integration, overall brand reputation and value protection. These key DD risk assessment outputs could make or break a deal.
Unfortunately, during the DD process there are many obstacles and biases that impact the validity of the results:
- Bias – Often the business stakeholder who identified the M&A opportunity may have already made the decision to buy the target entity based on revenue growth target or other drivers such as onshoring. So, there may be a bias in the decision to move forward despite any concerns raised by Quality & Regulatory that may be shortchanged because they threaten the deal.
- Timelines – while there are exceptions, the DD assessment period is only 3 – 6 weeks at the longest due to the urgency in making the M&A decision and minimize disruption of business management for both the M&A Target and the potential buyer and thereby “fish or cut bait”. This limited time impacts the ability to perform a thorough assessment.
- Prioritization – Usually the focus of the investment may be one significant product. What if other products that come along with the acquisition have significant regulatory or product quality vulnerabilities and are not examined during due diligence? This may lead to significant hidden cost to fix, drop from portfolio, and possibly brand damage.
- How deep can you go? – The confidentiality of the DD may limit the number of personnel that can be accessed and interviewed and transparency of the data collected.
- Expertise – Additionally, many Medical Device and Pharmaceutical companies will use their own employee functional representatives to perform the DD. However, they may be limited in their DD experience which may impact their ability to quickly identify significant Quality and Regulatory vulnerabilities.
All the above can compromise the DD output and may lead to underestimating the risks involved in acquiring the target entity as well as the cost to fix any inherent product quality and regulatory compliance shortcomings. Buyer entities should strongly consider augmenting their internal personnel functional team members with some outside Consultant expertise with broader DD experience and skills to overcome some of the obstacles cited above and provide credence to the assessment output. Often the Finance function will involve an Accounting / Financial Consulting firm, but we find that Quality and Regulatory functions could benefit from using experts in their fields as well.
A possible order of DD Quality and Regulatory assessment areas priority and duration are shown below:
- Verifying design robustness and design for manufacturing potential ~ 1 week
- Evaluating the FDA documentation and feasibility study data ~ 1 week
- Auditing the contract manufacturer and key Supply Chain partners for compliance, capacity, and risks ~1 week (2 days onsite + visit key suppliers)
- Interviewing the leadership team to assess their vision, capabilities, and alignment with company culture ~ 2 days
- Assessing the contractor/consultant’s processes and track record with QMS and regulatory activities ~1 week
- Identify potential staffing and infrastructure needs ~ 2 days
Should the decision be a “GO” to purchase the M&A target, it’s important that the risks are well documented and the cost soundly estimated to address any major product quality and regulatory compliance vulnerabilities and complete the integration of the Quality Systems. Again, using experienced Regulatory and Quality Consulting firms to assist with DD can leverage their vast experience to develop better estimates than otherwise and factor that cost into the price negotiation, escrow accounts, and/or post-merger reserve funds allocation.
Quality Systems Integration (QSI)
So, after completing your DD and the company has made the decision to acquire the new business, you don’t stop your investment there.
Now it’s time to invest in the new business’ product quality, its people, and QS infrastructure to ensure a long-term return from your purchase and to protect your brand image.
For the business and finance stakeholders in your organization, there may need to be a reminder of what kind of business you are in … A business that provides revenue and net income by providing high quality, medically necessary, products for patient care.
And who is your patient? It could be someone across the globe, your neighbor, a family member, or you. This is your Why. We are constituents and stakeholders of an amazing industry that everyday can make decisions and take actions could be a positive contribution to society or detrimental. Everyday … by the attitudes, behavior, and quality culture foundation that drive our behavior.
Nevertheless, not one size fits all for Quality Systems Integration (QSI). Depending on the organization and your business transition strategy you may fall under one of the following typical QSI scenarios listed in the table below along with some of the potential pros and cons:
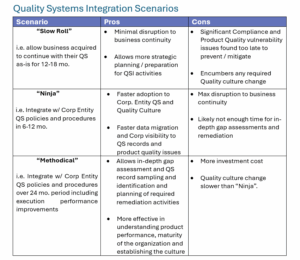
However, we have found more success with our clients using the “Methodical” approach as it allows for a more in-depth gap assessment and better identification of remediation activities that will pay dividends on product quality, adoption of the desired corporate quality culture, brand value, and long-term return on investment.
Here are five typical goals of Quality System Integration and how it differs from Due Diligence:

To be effective at QSI there should be a good program management infrastructure put in place such as shown below presented at the 2025 MedCon Conference:
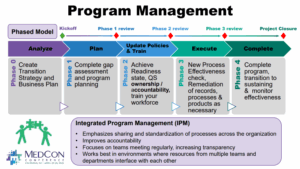
During the Analyze Phase the assessment priority of site(s) and products are established and aligned with the business goals on where to invest.
The Plan Phase involves establishing the Program plan (budget, resources, schedule) after completing the gap assessment and prioritizing any remediation required. The Gap Assessment usually involves the following steps and roles for Client vs. Consultant team members:
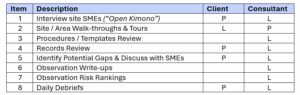
![]()
Next, the Update Policies and Training Phase will align the acquired business with corporate entity policies and address any QS shortcomings identified during the Gap Assessment. Ensuring there is accountability for each Ownership of each QS area is critical for success. Rarely do we find that procedures solely are the areas of deficiency. Instead, we find that organizations too often depend on Quality to be the police, and that regulatory compliance is their problem. However, we have found that QS Ownership belongs with the function that is most responsible for the execution of that system.
For example, R&D would be the most appropriate candidate to “own” Design Controls since it drives the majority of the activities required to be completed during product development, with the Quality function as one of many supporting functions. R&D would then be held accountable for the regulatory compliance effectiveness for Design Control, not Quality. Similarly, for “Process Validation”, where Operations and/or Engineering should be the Owners, and so forth.
During the Execution phase, after fully implementing the new QS subsystem(s), there should also be periodic effectiveness checks, conducted by team members independent of those generating the QS record outputs as well as any remediation records, to ensure they are meeting the intended quality required to satisfy regulatory compliance scrutiny. A defined audit checklist and sampling plan with established Confidence & Reliability specifications is recommended to validate that the outputs of the new systems are satisfactory.
To ensure efficiency and timely completion of all QSI deliverables, it is recommended to have Integrated Program Management (IPM). Typically, teams are established for each QS Workstream (ex. CAPA & NonConformances, P&PC, Process Validation, Design Control, Purchasing Controls, Complaint Handling, Risk Mgmt, Non-Product SW, etc.) with designation of a team lead and backup. Usually, the team will be comprised with some combination of full-time and part-time members familiar with sub-QS function and any special skills required (ex. Scientific, Engineering, IT, or other technical expertise). Team Leads work with IPM to develop project schedules, identify resource needs, report progress, and escalate issues as necessary to Executive Management to ensure obstacles are readily addressed to facilitate team success. Cross WS inputs and outputs are identified and tracked to completion as well as within WS deliverables completion.
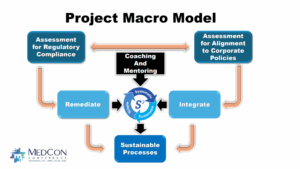
The Project Macro Model depicted below describes the flow of QSI phases and interactions with substantial Coaching and Mentoring of Team Members by Consultants along the journey required to successfully build sustainable QS processes.
About the Authors
Braulio Ortiz
With over 40 years of industry experience, Braulio Ortiz is the Director of Engineering, Quality Systems, and Operations at Baxter Healthcare. He is also the Principal and Founder of BioTeknica, Inc. Braulio holds a B.S. in Chemical Engineering from Cornell University and an MBA from the University of Miami. He is a recognized expert in regulatory compliance and engineering within the life sciences sector.
Dennett Kouri
Dennett Kouri brings more than 25 years of experience in the medical device and biologics industries. As the former Senior Vice President of Corporate Quality, Regulatory, and Clinical at Edwards Lifesciences, he led global regulatory compliance efforts and quality assurance activities in alliance management and due diligence. Dennett holds degrees from UC Santa Barbara and Southwestern University School of Law in Los Angeles.
Reference: Click the link to see the full MedCon 2025 presentation - MedCon Due Diligence QS Integration Presentation